JACS: A Novel MSH-Based Skeletal Editing Strategy for Pyridine-to-Pyridazine Transformation – Essential Synthetic Insights!

A carbon-to-nitrogen exchange-based skeletal editing method for converting pyridines to pyridazines has been successfully established. This approach employs MSH as the nitrogen source and utilizes mCPBA to promote ring remodeling. It operates under mild conditions, requires no inert atmosphere or light irradiation, and exhibits excellent functional group tolerance. Furthermore, the method has been applied to the late-stage modification of complex drug molecules, serving as a powerful tool for diversifying heterocycle chemistry and advancing drug discovery.
Pyridine compounds are widely used in the pharmaceutical field, yet their structural analogs, pyridazines—which feature two adjacent ring nitrogen atoms—have long been underexplored due to limitations in synthetic routes. This work introduces a skeletal editing strategy that enables the conversion of pyridines into pyridazines by replacing one carbon atom in the pyridine ring with a nitrogen atom while preserving aromaticity. The specific pathway employs MSH as an electrophilic nitrogen source, mCPBA as an oxidant, and Cs₂CO₃ as a base, accomplishing the transformation via a two-step reaction at room temperature (see Figure c for detailed procedures). This method is straightforward, operates under mild conditions, exhibits a broad substrate scope, and is compatible with complex drug molecules, offering a novel tool for expanding heterocycle chemical space and optimizing drug candidates at a late stage.
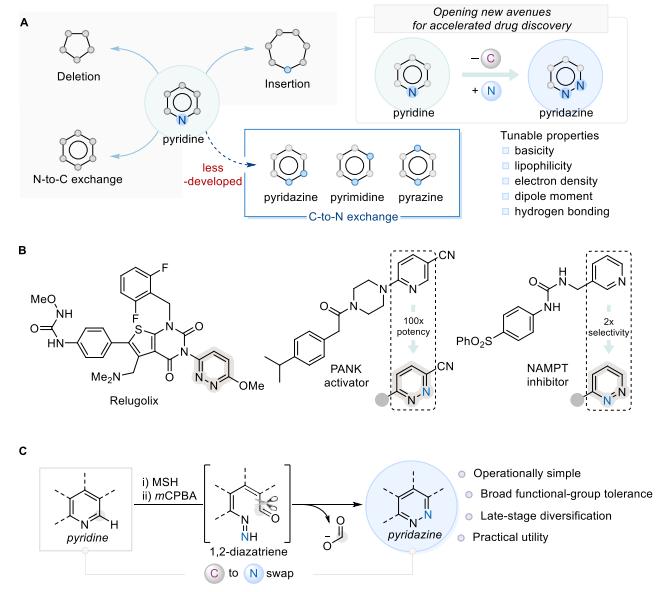
Drawbacks of Conventional Methods: The Levin group previously reported a method for synthesizing pyridazines starting from 2-chloropyridine. While valuable, this approach requires additional steps to access the 2-chloropyridine precursor, including mCPBA oxidation of pyridine followed by POCl₃-mediated chlorination. Moreover, the method necessitates the presence of an electron-withdrawing substituent on the pyridine ring and must be conducted under near-UV light (390 nm) irradiation. It also generates toxic by-products (HN₃ and hydrocyanic acid), thereby hindering structure–activity relationship studies in drug research.

I. Skeletal Editing from Pyridine to Pyridazine
1. Reaction Conditions
The transformation proceeds via a straightforward two-step procedure: N-amination of pyridine using O-mesitylenesulfonylhydroxylamine (MSH) as an electrophilic nitrogen source, followed by carbon–nitrogen bond formation through a 1,2-diazetine intermediate. This step is mediated by m-chloroperoxybenzoic acid (mCPBA, 2.3 equiv) as the oxidant and cesium carbonate (Cs₂CO₃, 3.0 equiv) as the base under ambient air at room temperature. The method requires no UV light irradiation or pre-installed directing groups.
2. Optimal Reaction Parameters
l Solvent: DMF is preferred, but tetrahydropyran (THP) can be used as an alternative due to its ease of removal.
l Oxidant Loading: mCPBA is used at 2.3 equivalents, with 1 equivalent dedicated to ring remodeling, 1 equivalent driving the Baeyer–Villiger oxidation, and 0.3 equivalents compensating for minor consumption. Yields decrease significantly below 2 equivalents, and no notable improvement is observed beyond 2.3 equivalents.
l Essential Reagents: Both Cs₂CO₃ and mCPBA are indispensable; omission of either prevents formation of the target product.
l Reaction Atmosphere: The process proceeds efficiently at room temperature under air, requiring no inert gas protection or specialized temperature control.

II. Substrate Scope and Applications
1. Substrate Scope
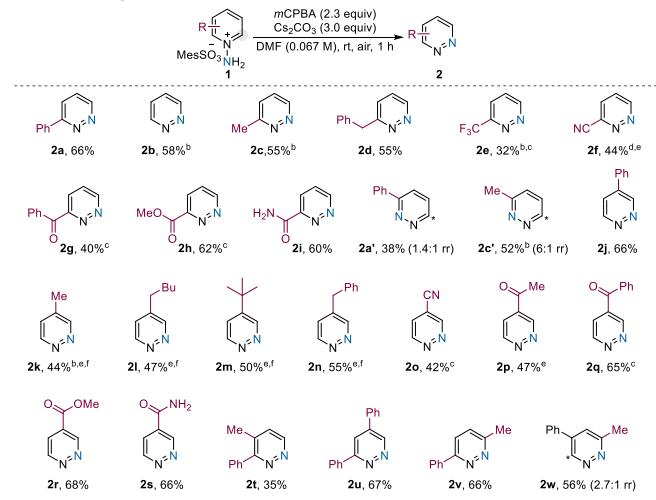
The method exhibits broad generality and functional group tolerance, accommodating both electron-rich and electron-deficient systems, with most reactions completing within one hour. It demonstrates excellent compatibility with ketones, thiophenes, pyridyl groups, and free alcohols.
l C2-Substituted Pyridines
All C2-substituted pyridines undergo smooth conversion, including aryl (2a), unsubstituted (2b), and alkyl (2c/2d) variants. Substrates bearing electron-withdrawing groups such as trifluoromethyl (2e) and cyano (2f), as well as carbonyl-containing functionalities including benzoyl (2g), ester (2h), and amide (2i), are all efficiently transformed.
l C3-Substituted Pyridines
Methyl-substituted substrate (2c′) reacts preferentially at the C2 position with a regioselectivity ratio of C2:C6 = 6:1. For phenyl-substituted substrate (2a′), steric hindrance reduces regioselectivity to C2:C6 = 1.4:1.
l C4-Substituted Pyridines
Long alkyl chains (2l) and sterically demanding groups such as tert-butyl (2m) are well tolerated, yielding products with efficiency comparable to C2-substituted analogues.
l Polysubstituted and Heteroaryl-Fused Pyridines
Polysubstituted substrates (2t–2w) and pyridines bearing oxidatively sensitive heteroarenes such as thiophene (2x) and pyridyl (2y) undergo highly selective conversion, generating diverse pyridazine products (2x–2ab).

2. Applications
This method enables direct skeletal editing of structurally complex marketed drug molecules, efficiently generating their pyridazine analogues, and has been successfully demonstrated on a gram scale, confirming its practical utility. Furthermore, the resulting pyridazine core can be further transformed into the biologically relevant triazolo[4,3-b]pyridazine scaffold, underscoring its synthetic value in medicinal chemistry. Pyridazines offer distinct advantages for drug discovery: compared to pyridine, they exhibit lower basicity, reduced lipophilicity, stronger electron-deficient character, higher dipole moment, and two hydrogen-bond acceptors.

Bisacodyl was converted to its pyridazine analogue (2ad), and loratadine was modified to incorporate the pyridazine core (2ae). Additionally, an olefin-containing donepezil derivative (2af) and the anticholinergic agent tropicamide (2ag) also underwent smooth conversion.
III. Mechanistic Validation
Density functional theory (DFT) calculations support the proposed mechanism: mCPBA attacks the pyridine ring to form a dearomatized adduct, which subsequently undergoes ring opening, 6π-electrocyclization, and oxidative carbon extrusion to restore aromaticity.

IV. Conclusion
A novel skeletal editing strategy has been developed that efficiently converts pyridines to pyridazines via a carbon-to-nitrogen exchange. The sequence involves N-amination followed by mCPBA-promoted ring remodeling. Operating under mild conditions with broad substrate scope and high functional group compatibility, this method enables precise skeletal editing of complex drug molecules. This work addresses a long-standing challenge in synthetic chemistry and significantly expands the toolbox for heterocycle diversification in medicinal chemistry.
Read More
https://pubs.acs.org/doi/10.1021/jacs.5c15601